近日,中国工程院院士、太阳集团8722c齐鲁医院络病理论创新转化全国重点实验室张运教授,欧洲科学院院士张澄教授和太阳集团8722c齐鲁医院心血管内科张文程教授团队在心血管重构基础研究领域中,发现了多个干预新靶点,在Nature旗下Signal Transduction and Targeted Therapy(中科院1区,最新影响因子40.8)、Advanced Science(中科院1区,最新影响因子14.3)、Chemical Engineering Journal(中科院1区,最新影响因子13.3)发表系列论文,受到国际学术界的高度关注。
阿霉素是治疗多种恶性肿瘤的有效药物,进入临床应用已50年,但此药具有损伤组织器官的副作用,尤其是阿霉素心肌病,可导致心力衰竭和死亡。然而,阿霉素心肌病的发病机制不明,治疗靶点不清。目前阿霉素心肌病的临床治疗主要针对已发生的心力衰竭,尚无药物可预防阿霉素的心脏毒性。因此,迫切需要明确阿霉素心肌病的发生机制并寻找有效的干预靶点。
解联蛋白和金属蛋白酶17(ADAM17)是一个由824个氨基酸组成的跨膜蛋白。本实验的以往研究表明,ADAM17在多种心血管疾病的发病中扮演了重要角色。近年研究发现,接受阿霉素化疗的临床患者和实验小鼠中肿瘤坏死因子α(TNF-α)的血清水平显著上升,且阿霉素治疗的小鼠心肌组织中TNF-α表达亦显著增加。由于ADAM17可通过剪切作用将tmTNF-α转化为sTNF-α,后者可发挥生物学作用,因此ADAM17又被称为肿瘤坏死因子转化酶。然而,ADAM17和TNF-α在阿霉素心肌病中的作用尚不明了。
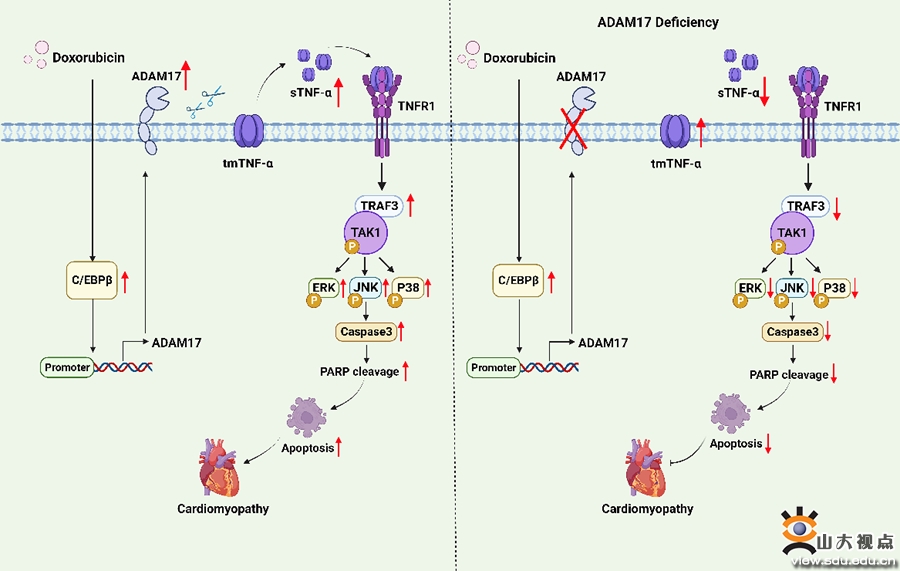
张运院士、张澄院士和杨建民教授课题组研究发现,在阿霉素心肌病小鼠心肌组织中,ADAM17的蛋白表达和活性显著升高,且主要在心肌细胞中表达。为明确心肌细胞ADAM17在阿霉素心肌病中的作用,课题组构建了心肌细胞特异性敲除ADAM17基因的小鼠模型和心肌细胞特异性过表达ADAM17的小鼠模型。在阿霉素心肌病小鼠模型中,明确了心肌细胞ADAM17敲除可减轻小鼠心肌重构、心肌萎缩及细胞凋亡水平,而心肌细胞ADAM17过表达则加剧了上述病理改变。为明确心肌细胞ADAM17影响阿霉素心肌病的机制,课题组使用小鼠心脏组织进行生物信息学分析,发现TNF信号通路在阿霉素心肌病及其对照组小鼠的心脏组织之间存在显著差异。深入研究发现,心肌细胞ADAM17通过促进可溶性TNF-α脱落,上调TRAF3的表达。增加的TRAF3与TAK1结合,促进TAK1自磷酸化并活化,导致下游MAPK通路被激活,进一步促进心肌细胞凋亡,最终加速了阿霉素心肌病的进展。此外。课题组证明,在乳腺癌和黑色素细胞瘤的小鼠模型中,心肌细胞ADAM17基因敲除不仅对阿霉素心肌病具有心脏保护作用,而且不影响阿霉素的抗肿瘤作用。总之,这些发现可能为阿霉素心肌病提供一个新的预防和治疗靶点。该研究近日发表于Signal Transduction and Targeted Therapy,太阳集团8722c齐鲁医院心内科张运院士、张澄院士和杨建民教授为该文的共同通讯作者,太阳集团8722c齐鲁医院心内科博士研究生谢琳和博士后薛飞为该文的共同第一作者,太阳集团8722c齐鲁医院为该文的第一和通讯作者单位。该研究成果已被iNature、iPubpeers、365医学网、代谢与整合生物学、课题指南针等国内多家公众号及媒体报道。产生了重大的学术影响。
根据世界卫生组织的统计,全球约10亿人患有高血压,每年约有900万人死于高血压并发症。因此,深入研究高血压的发病机制并寻找有效的干预手段具有重大的临床意义。基因的转录后调控尤其是mRNA的稳定性是调节基因表达的重要步骤,mRNA稳定性失调会直接导致包括编码生长因子、炎症细胞因子以及原癌基因等多种基因的异常表达,是多种疾病的发病基础。ZFP36是一个包含串联Cys-Cys-Cys-His (CCCH)锌指结构域的RNA结合蛋白,可通过与靶基因mRNA 3’非翻译区富含AU元件的区域结合,降低mRNA的稳定性。ZFP36在巨噬细胞、心肌细胞、肿瘤细胞等多种细胞中发挥重要作用,但在平滑肌细胞尤其是高血压发病中的作用尚不明了。
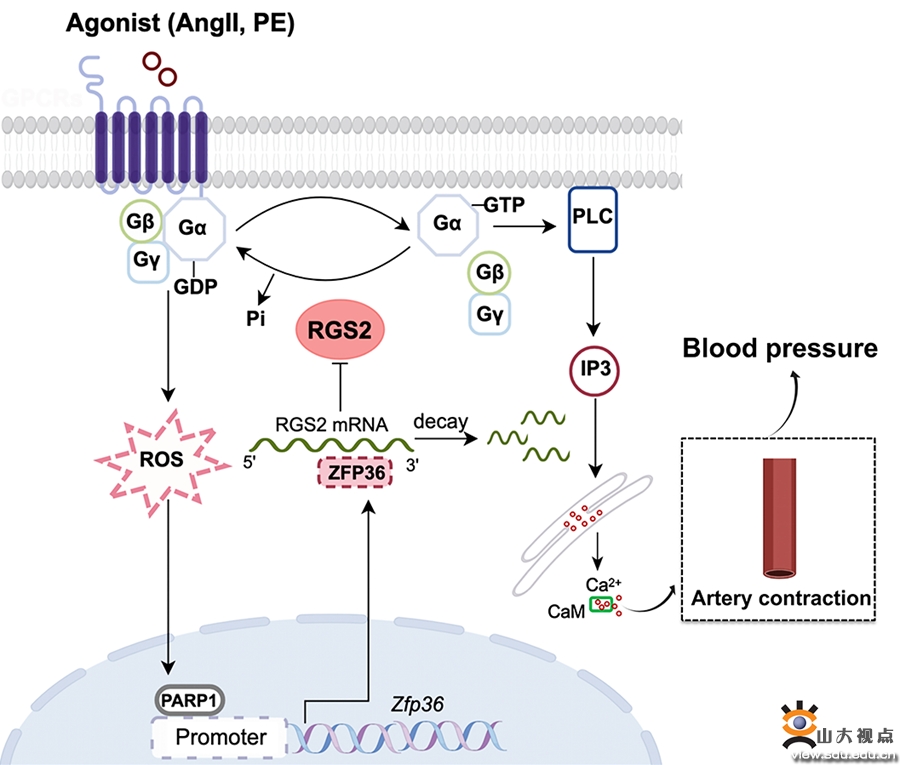
张文程教授课题组研究发现,ZFP36在高血压患者和高血压动物模型的血管平滑肌中表达增加,提示ZFP36可能参与高血压的发生和发展。课题组构建了平滑肌细胞特异性ZFP36敲除(Zfp36SMKO)小鼠,发现与对照鼠相比,Zfp36SMKO小鼠的收缩压和平均动脉压均下降。血管张力实验表明,与对照鼠相比,Zfp36SMKO小鼠的肠系膜动脉对NE、AngII、U46619及ET-1刺激引起的收缩反应均降低。深入的机制研究表明,RGS2是ZFP36调控血管收缩的靶分子, ZFP36通过结合并促进RGS2mRNA的降解抑制其蛋白表达,从而增加GPCRs通路介导的细胞内Ca2+水平,促进平滑肌收缩,维持血压稳定。体内研究表明,抑制RGS2能够逆转ZFP36敲除引起的小鼠血压下降。此外,平滑肌细胞敲除ZFP36可抑制AngII诱导的小鼠高血压及其血管重构。同时,ZFP36基因敲低显著降低了自发性高血压大鼠的收缩压和平均动脉压。总之,本研究揭示了ZFP36调控血管收缩和血压稳定的重要作用及分子机制,为抗高血压药物的开发提供了新的思路和靶点。该研究近日发表于Advanced Science杂志(中科院1区,最新影响因子14.3),张文程教授为该文的通讯作者,齐鲁医院心血管内科硕士研究生崔秀茹为该文的第一作者,太阳集团8722c齐鲁医院为该文的第一和通讯作者单位。
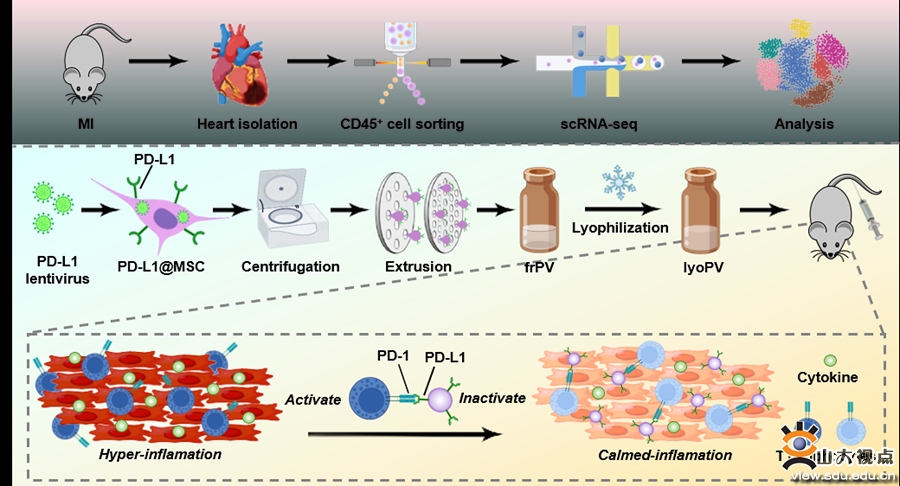
心肌梗死(MI)是全球心血管病死亡的主要原因。机体对于心肌损伤的应激免疫反应对于损伤组织的内源性修复和组织稳态至关重要。然而,大面积的组织坏死可能引发持续性炎症反应,降低机体对免疫系统的耐受性而诱导心室重构。研究发现,CD8杀伤性T淋巴细胞在缺血心肌中被动员和激活,释放颗粒酶B和干扰素-γ等炎性介质。因此,清除致病性CD8 T淋巴细胞有可能抑制过度炎症反应并缓解心脏损害。已知T淋巴细胞表达与抑制配体相互作用的免疫检查点对于维持自我耐受和预防自身免疫至关重要。免疫检查点PD-1与PD-L1之间的相互作用可诱导T细胞耗竭与失能,从而减轻自身和有害免疫的病理反应。因此,引入PD-L1分子有望激活PD-1/PD-L1抑制轴,进而减轻由T细胞驱动的心脏炎症。
为了明确T淋巴细胞在心肌梗死发展中的作用,张文程教授课题组联合苏州大学心血管病研究所沈振亚教授和陈维倩教授课题组,首先对小鼠梗死心脏的单细胞测序数据进行了详尽分析,揭示了T淋巴细胞群体的高度异质性以及增殖性CD8细胞毒性T淋巴细胞亚群的存在。鉴于传统基于分泌组的细胞外囊泡治疗策略常受限于稳定性不足和成本效益低下,联合课题组创新性地开发了一种室温稳定的、富含程序性细胞死亡配体1(PD-L1)的细胞膜纳米囊泡(PD-L1@NV),旨在减轻T细胞诱导的缺血性炎症反应。功能研究表明,PD-L1@NV展现出与T细胞膜的高效结合能力,抑制其增殖,并促进活化的CD8 T淋巴细胞耗竭,进而促进损伤心肌组织的修复。机制研究发现,PD-L1@NV通过减少CD8细胞毒性T淋巴细胞数量并增加调节性T细胞比例,重塑了体内T细胞的动态平衡。本研究开发的室温稳定型PD-L1@NV可针对T细胞异常活化诱发的免疫病态,为心肌梗死的治疗提供了一种简便、安全的策略。该研究近日发表于发表在Chemical Engineering Journal杂志(中科院1区,最新影响因子13.3),张文程教授为该文的共同通讯作者。
文章链接:
https://pubmed.ncbi.nlm.nih.gov/39406701/
https://pubmed.ncbi.nlm.nih.gov/39589932/
https://www.sciencedirect.com/science/article/abs/pii/S1385894724085218